
Διαβαζουμε στο Circulation Research που εχει Δείκτη απήχησης 16, από τους υψηλότερους δείκτες στο σύνολο των επιστημονικών περιοδικών παγκοσμίως…αλλα και στην Αμερικανικη Ιατρικη βιβλιοθηκη…Αμφισβητηστε αν μπορειτε οτι τα Εμβολια του κοβιντ ειναι η πορτα ωστε η ανθρωποτητα να οδηγηθει στα γονατα !!…Μιληστε ΣΗΜΕΡΑ γιατι καθημερινα οδηγειτε στο θανατο 1000δες ανθρωπους !
Ζητείται εισαγγελεας να σταματησει την ΓΕΝΟΚΤΟΝΙΑ των Ελληνων !
SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2
SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) infection relies on the binding of S protein (Spike glycoprotein) to ACE (angiotensin-converting enzyme) 2 in the host cells. Vascular endothelium can be infected by SARS-CoV-2,1 which triggers mitochondrial reactive oxygen species production and glycolytic shift.2 Paradoxically, ACE2 is protective in the cardiovascular system, and SARS-CoV-1 S protein promotes lung injury by decreasing the level of ACE2 in the infected lungs.3 In the current study, we show that S protein alone can damage vascular endothelial cells (ECs) by downregulating ACE2 and consequently inhibiting mitochondrial function.
We administered a pseudovirus expressing S protein (Pseu-Spike) to Syrian hamsters intratracheally. Lung damage was apparent in animals receiving Pseu-Spike, revealed by thickening of the alveolar septa and increased infiltration of mononuclear cells (Figure [A]). AMPK (AMP-activated protein kinase) phosphorylates ACE2 Ser-680, MDM2 (murine double minute 2) ubiquitinates ACE2 Lys-788, and crosstalk between AMPK and MDM2 determines the ACE2 level.4 In the damaged lungs, levels of pAMPK (phospho-AMPK), pACE2 (phospho-ACE2), and ACE2 decreased but those of MDM2 increased (Figure [B], i). Furthermore, complementary increased and decreased phosphorylation of eNOS (endothelial NO synthase) Thr-494 and Ser-1176 indicated impaired eNOS activity. These changes of pACE2, ACE2, MDM2 expression, and AMPK activity in endothelium were recapitulated by in vitro experiments using pulmonary arterial ECs infected with Pseu-Spike which was rescued by treatment with N-acetyl-L-cysteine, a reactive oxygen species inhibitor (Figure [B], ii).
Figure.
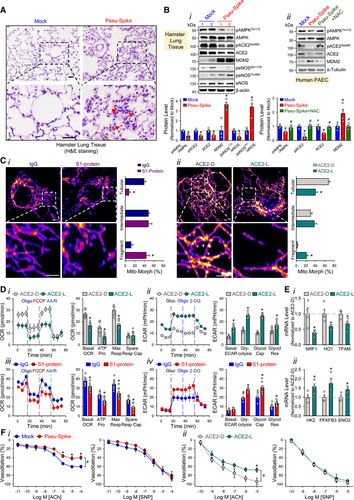
Figure. SARS-CoV-2 (Severe acute respiratory syndrome coronavirus 2) Spike protein exacerbates endothelial cell (EC) function via ACE (angiotensin-converting enzyme) 2 downregulation and mitochondrial impairment.A, Representative H&E histopathology of lung specimens from 8- to 12 wk-old male Syrian hamsters 5-day post administration of pseudovirus overexpressing Spike protein (Pseu-Spike) or mock virus in control group (n=3 mice per group, 1×108 PFU). Thickened alveolar septa (red arrowhead) and mononuclear cell (red arrow). Scale bar=20 μm. B, Pseu-Spike (n=4) or mock virus (n=4)–infected hamster lungs were subjected to Western blot analysis for pAMPK (phospho-AMPK) T172, AMPK, pACE2 (phospho angiotensin-converting enzyme) S680, ACE 2, MDM2, peNOS S1176, peNOS T494, eNOS (endothelial NO synthase), and β-actin (B, i). Human pulmonary arterial EC (PAECs) were infected with Pseu-Spike or mock virus for 24 h with or without N-acetyl-L-cysteine (NAC; 5 mmol/L) pretreatment for 2 h. The protein extracts were analyzed by Western blot using antibodies against proteins as indicated (n=4; B, ii). C, Representative confocal images of mitochondrial morphology of ECs treated with human recombinant S1 protein or IgG (4 μg/mL) for 24 h (C, i) or infected with human adenovirus ACE2 S680D (ACE2-D) or ACE2 S680L (ACE2-L; 10 MOI) for 48 h (C, ii). Mitochondria were visualized using TOM20 antibody (n=4, 50 cells counted for each replicate). Scale bar=2.5 μm. Tubular: the majority of mitochondria in ECs was >10 μm in length; Intermediate: the mitochondria were <≈10 μm; Fragment: the majority of mitochondria were spherical (no clear length or width). D, Measurement of oxygen consumption rate (OCR, D, i and iii) and extracellular acidification rate (ECAR, D, ii and iv) in ECs infected with ACE2-D vs ACE2-L (10 MOI) for 48 h (n=3) or treated with IgG vs S1 protein (4 μg/mL) for 24 h (n=3). E, Real-time quantitative polymerase chain reaction analysis of the indicated mRNA levels in lung ECs from ACE2-D (n=4) and ACE2-L (n=4) knock-in mice. Eight-week-old ACE2-D and ACE2-L male mice with C57BL/6 background were used. F, Dose-response curves of acetylcholine (ACh, left)- and sodium nitroprusside (SNP, right)–mediated relaxation on the tension of phenylephrine (1 μmol/L) precontracted intrapulmonary artery stripes from Pseu-Spike-(ACh n=8, SNP n=5) or mock (ACh n=6, SNP n=5) virus–infected Syrian hamsters (1×108 PFU; F, i) and ACE2-D (n=6) or ACE2-L (n=5) mice (F, ii). The animal experiments were approved by the ethical committee of Xi’an Jiaotong University. 2-DG indicates 2-Deoxy-D-glucose; ACE2-D, a phospho-mimetic ACE2 with increased stability; ACE2-L, a dephospho-mimetic ACE2 with decreased stability; AMPK, AMP-activated protein kinase; AA/R, antimycin A&Rotenone; ENO2, enolase 2; FCCP, carbonyl cyanide-p-(trifluoromethoxy)phenylhydrazone; H&E, Hematoxylin and Eosin; HK2, hexokinase 2; HO1, heme oxygenase-1; MDM2, murine double minute 2; MOI, multiplicity of infection; NRF1, nuclear respiratory factor 1; peNOS, phospho-eNOS; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; Resp, respiration; and TFAM, transcription factor A, mitochondrial. We next studied the impact of S protein on mitochondrial function. Confocal images of ECs treated with S1 protein revealed increased mitochondrial fragmentation, indicating altered mitochondrial dynamics (Figure [C], i). To examine whether these mitochondrial changes were due, in part, to the decreased amount of ACE2, we overexpressed ACE2 S680D (ACE2-D, a phospho-mimetic ACE2 with increased stability) or S680L (ACE2-L, a dephospho-mimetic with decreased stability)4 in ECs. As shown in Figure [C], ii, ECs with ACE2-L had a higher number of fragmented mitochondria when compared to those with ACE2-D. Performing oxygen consumption rate and extracellular acidification rate assays, we found that ECs overexpressing ACE2-L had reduced basal mitochondrial respiration, ATP production, and maximal respiration compared to ECs overexpressing ACE2-D (Figure [D], i). Moreover, ACE2-L overexpression caused increased basal acidification rate, glucose-induced glycolysis, maximal glycolytic capacity, and glycolytic reserve (Figure [D], ii). Also, ECs incubated with S1 protein had attenuated mitochondrial function but increased glycolysis, when compared with control cells treated with IgG (Figure [D], iii and iv). We also compared the expressions of mitochondria- and glycolysis-related genes in lung ECs isolated from ACE2-D or ACE2-L knock-in mice.4 Shown in Figure [E], the mRNA levels of NRF1, HO1, and TFAM (mitochondria biogenesis-related genes) were increased, whereas those of HK2, PFKFB3, and ENO2 (glycolysis-related genes) were decreased in lung ECs in ACE2-D mice, as compared to those in ACE2-L mice. SARS-CoV-2 infection induces EC inflammation, leading to endotheliitis.1,5 Because S protein decreased ACE2 level and impaired NO bioavailability, we examined whether S protein entry is indispensable for dysfunctional endothelium. As shown in Figure [F], i, the endothelium-dependent vasodilation induced by acetylcholine was impaired in pulmonary arteries isolated from Pseu-Spike-administered hamsters, whereas the endothelium-independent vasodilation induced by sodium nitroprusside was not affected. We also compared the acetylcholine- and sodium nitroprusside–induced vasodilation of pulmonary vessels from ACE2-D or ACE2-L mice. As anticipated, acetylcholine-induced vasodilation was hindered in pulmonary arteries isolated from ACE2-L mice in comparison to ACE2-D mice (Figure [F], ii). There was, however, little difference in sodium nitroprusside–induced vasodilation between ACE2-D and ACE-L animals. Although the use of a noninfectious pseudovirus is a limitation to this study, our data reveals that S protein alone can damage endothelium, manifested by impaired mitochondrial function and eNOS activity but increased glycolysis. It appears that S protein in ECs increases redox stress which may lead to AMPK deactivation, MDM2 upregulation, and ultimately ACE2 destabilization.4 Although these findings need to be confirmed with the SARS-CoV-2 virus in the future study, it seems paradoxical that ACE2 reduction by S protein would decrease the virus infectivity, thereby protecting endothelium. However, a dysregulated renin-angiotensin system due to ACE2 reduction may exacerbate endothelial dysfunction, leading to endotheliitis. Collectively, our results suggest that the S protein-exerted EC damage overrides the decreased virus infectivity. This conclusion suggests that vaccination-generated antibody and/or exogenous antibody against S protein not only protects the host from SARS-CoV-2 infectivity but also inhibits S protein-imposed endothelial injury. Nonstandard Abbreviation and Acronyms ACE angiotensin-converting enzyme ECs endothelial cells eNOS endothelial NO synthase pACE2 phospho-ACE2 pAMPK phospho-AMPK S protein Spike glycoprotein Data Availability The data that support the findings of this study, including statistical analyses and reagents used, are available from the corresponding author upon request. Sources of Funding This work was supported in part by grants from the National Natural Science Foundation of China (NSFC) grants 81870220 (S. Wang), 81800328 (J.Z.), 81941005 (Z.-Y. Yuan); Shaanxi Natural Science Fund S2020-JC-JQ-0239 (S. Wang); The National Key Research and Development Program (Grant No. 2018YFC1311500; Z.-Y. Yuan); the Clinical Research Award of the First Affiliated Hospital of Xi’an Jiaotong University (Grant No. XJTU1AF-CRF-2016-004; Z.-Y. Yuan); Xi’an Jiaotong University Financial support. Disclosures None. Footnotes *Y. Lei and J. Zhang contributed equally. †U. Manor, S. Wang, Z.-Y. Yuan, and J.Y.-J. Shyy contributed equally as senior authors. For Sources of Funding and Disclosures, see page 1324. Correspondence to: John Y-J. Shyy, PhD, Division of Cardiology, Department of Medicine, University of California, San Diego, 9500 Gilman Dr, La Jolla, CA 92093, Email [email protected] Zu-Yi Yuan, MD, PhD, Department of Cardiology, First Affiliated Hospital of Xi’an Jiaotong University, 277 Yanta W Rd, Xi’an 710061, China, Email [email protected] https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.121.318902
Να δουμε και τι εχει μεσα η Αμερικανικη ιατρικη βιβλιοθηκη; ..Για να μην εχει κανεις την θεση της αντινοησης για οποιοδηποτε θεμα; ..Εδω λοιπον https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8091897/
Εχουμε μηπως να πουμε οτι καποιος λεει ψεματα ; Οτι καποιοι ειναι γραφικοι ;Θα βρουμε καποιο τροπο να αποδομησουμε την αποψη των ερευνων απο επιστημονες ;;..Μηπως πρεπει να χωρισουμε τους επιστημονες σε μαυρους και ασπρους που συμφωνουν η διαφωνουν με την γενοκτονια των ανθρωπων ;
Απαντηστε αν τολματε , Που ειναι οι δημοσιογραφοι να μιλησουν ;..Οταν ερθει η στιγμη της απολογιας δεν θα εχετε να πειτε τιποτα ..Γιατι εχετε ηδη ενημερωθει!!!…

{kind=link}